This is an old revision of the document!
Introduction to Galaxy
| Instructor | Stephanie Le Gras |
|---|---|
| Duration | 3.5 hours |
| Content | Description of the key features of Galaxy (Lecture) |
| Practical session on basic features of Galaxy (Hands-on) | |
| Prerequisites | None |
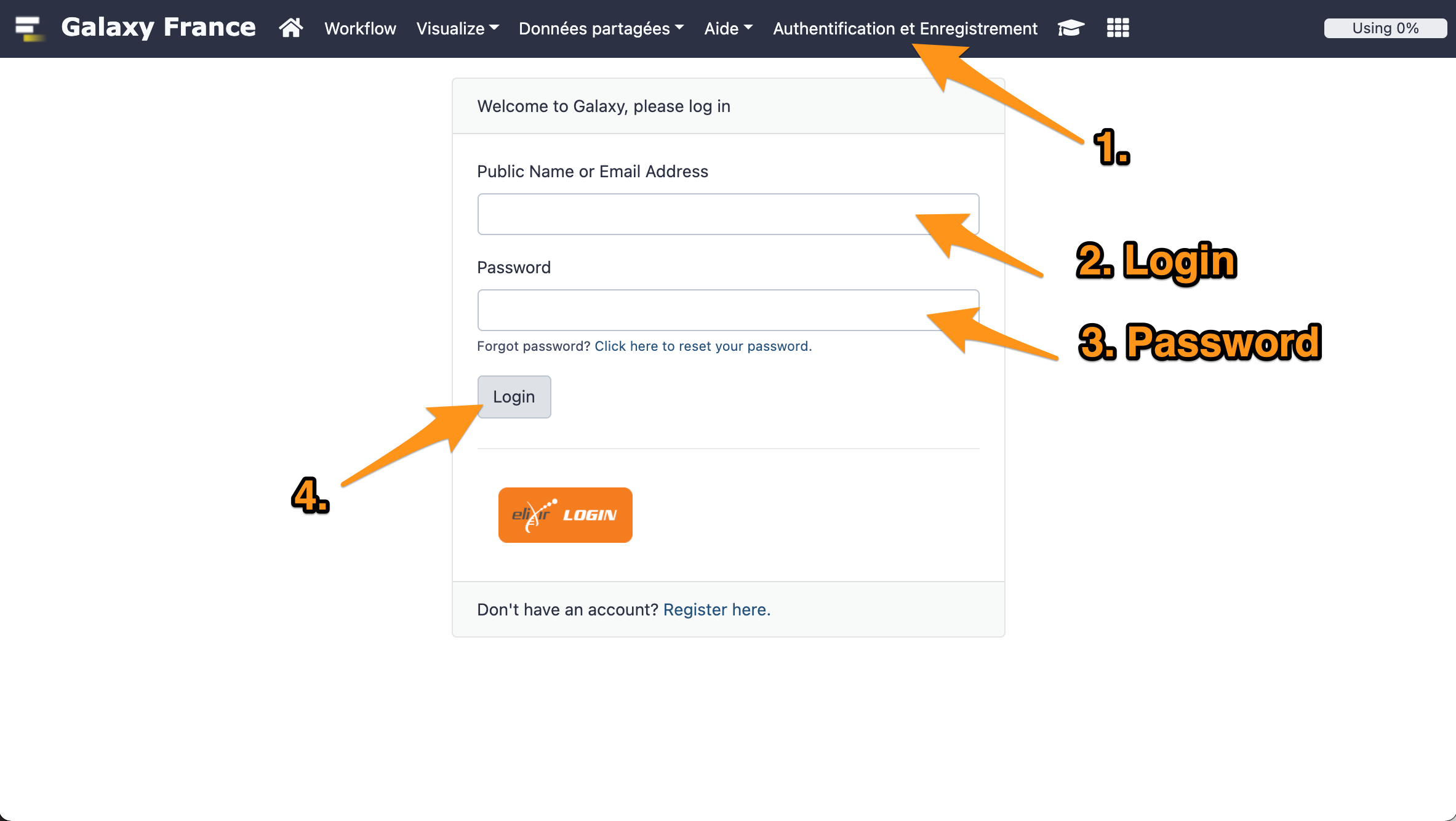
10 Log in to GalaxEast
- Go to Galaxy France website
- Log in with your account.
11 History
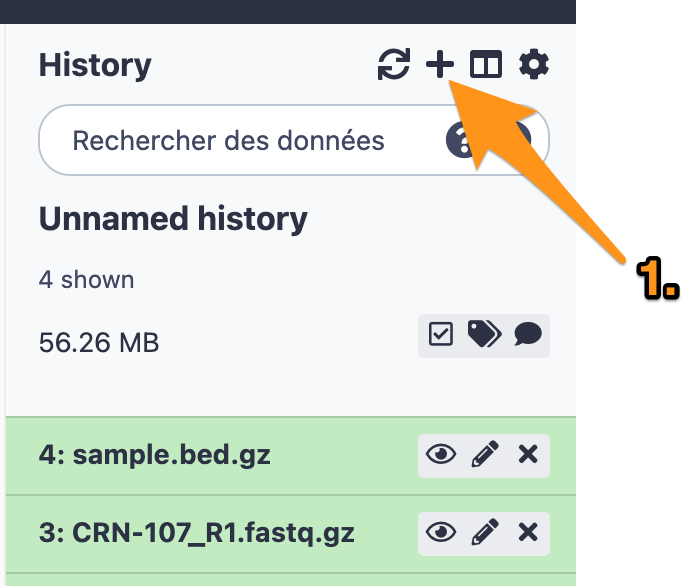
11.1 Create a new history
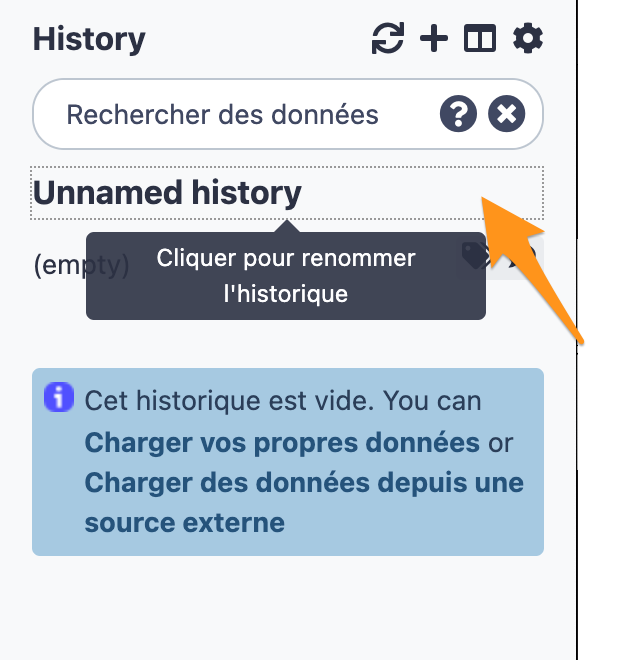
11.2 Change the name of the history to “DNA-seq data analysis"
12 Import data into Galaxy
12.1 Import files from your computer to Galaxy
12.2 Import a file from your computer
- Download the file “sample.bed.gz” following this link and upload it to Galaxy.
- The genome is: Mouse (mm9)
- The format is: bed
13 Remove a dataset
- Remove the dataset sample.bed from your history by clicking on the button
- You are told that your history is empty. Look at the size of your history
- Click on “deleted” in the top of the history panel (below the history name). Remove definitely the file from the disk by clicking on “Permanently remove it from disk”.
- Click on “hide deleted”
14 Running a tool
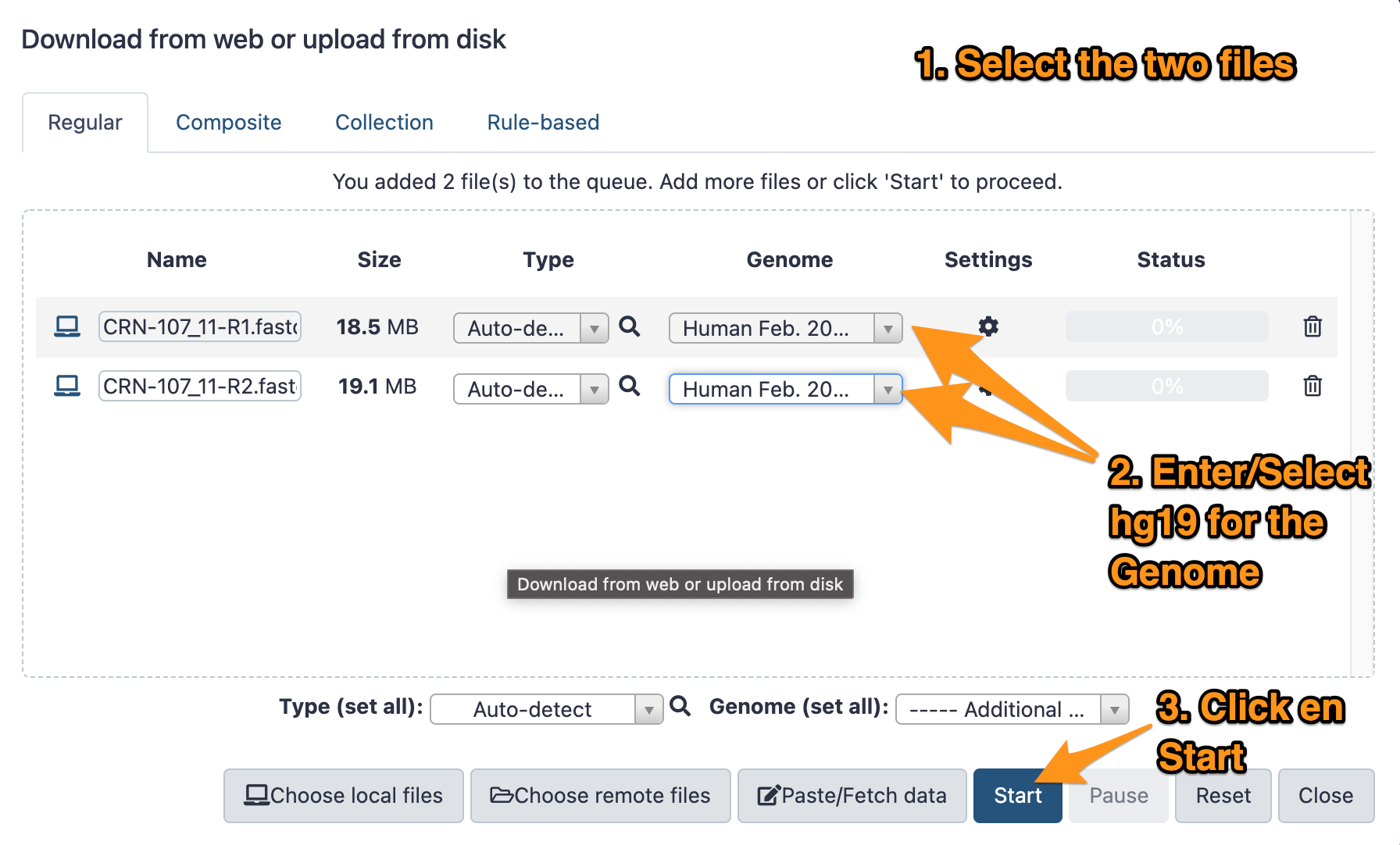
- Download the two files CRN-107_11-R1.fastq.gz and CRN-107_11-R2.fastq.gz following this link.
- Import them to your history called “DNA-seq data analysis”
- The genome is: Human (hg19)
- The format: <auto detect>
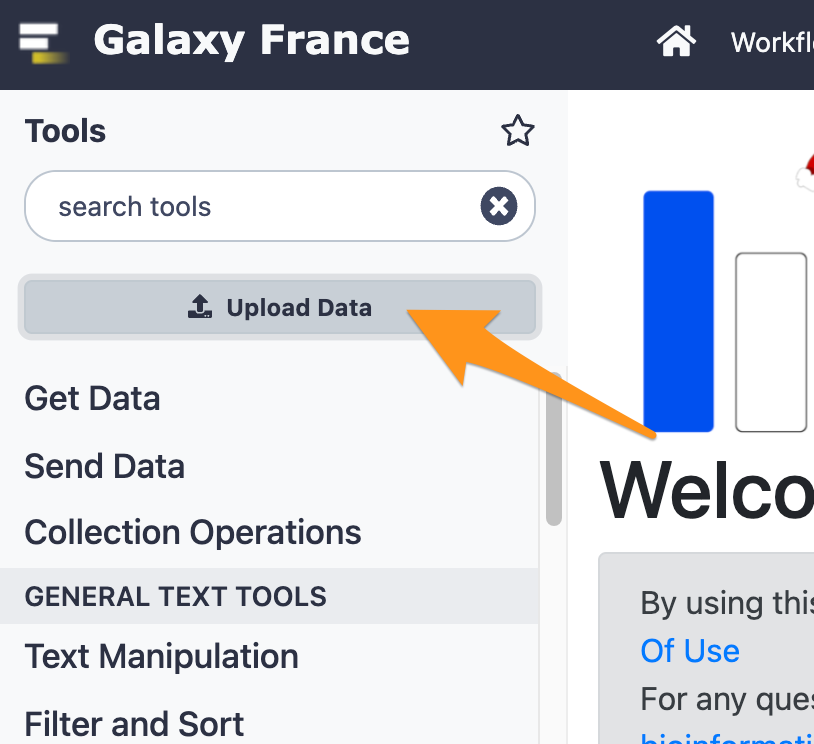
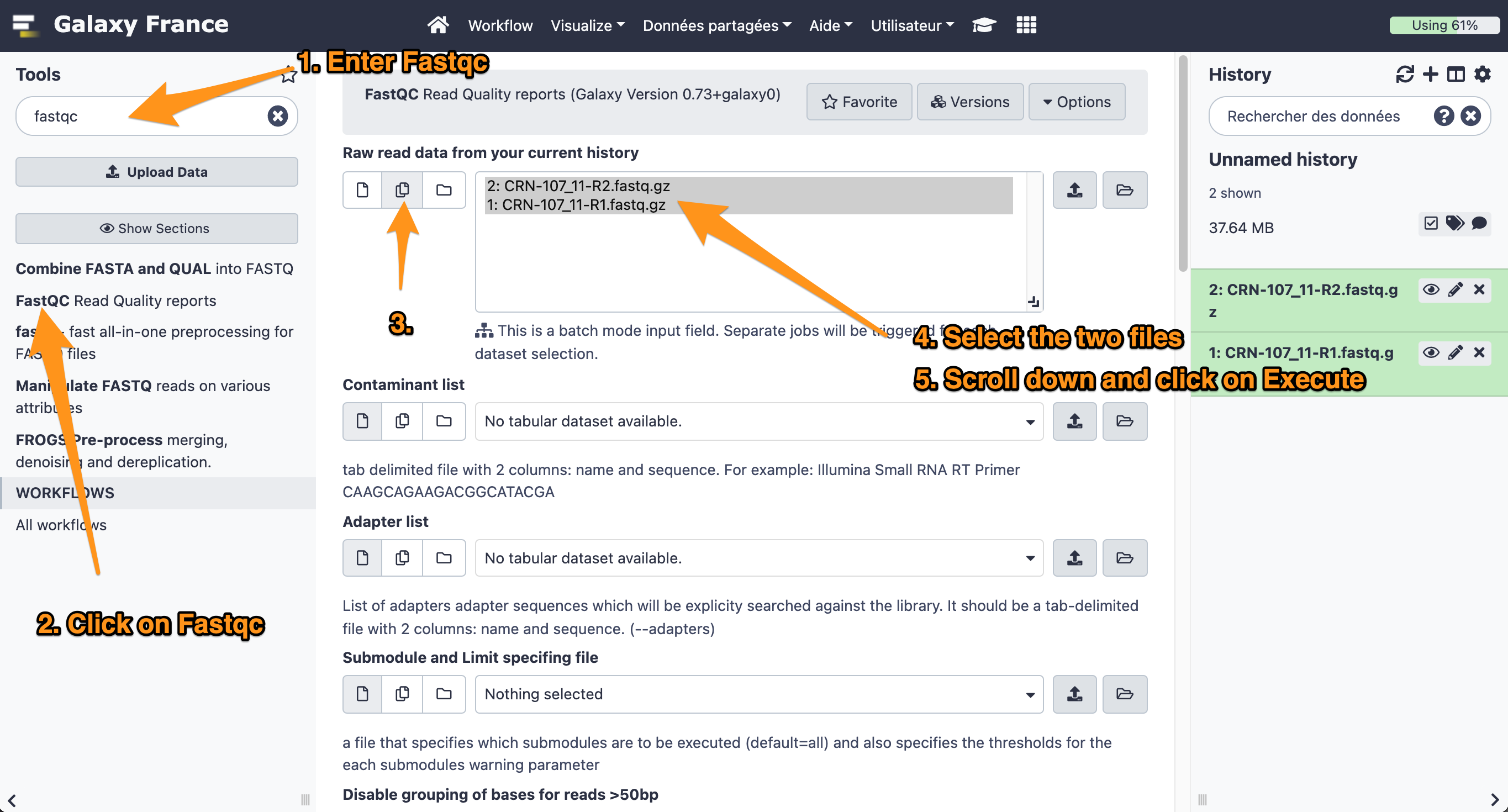
- Use the tool “FastQC Read Quality reports” to compute quality analysis on the datasets “CRN-107_11-R1.fastq” and “CRN-107_11-R2.fastq”
- Use default parameters.
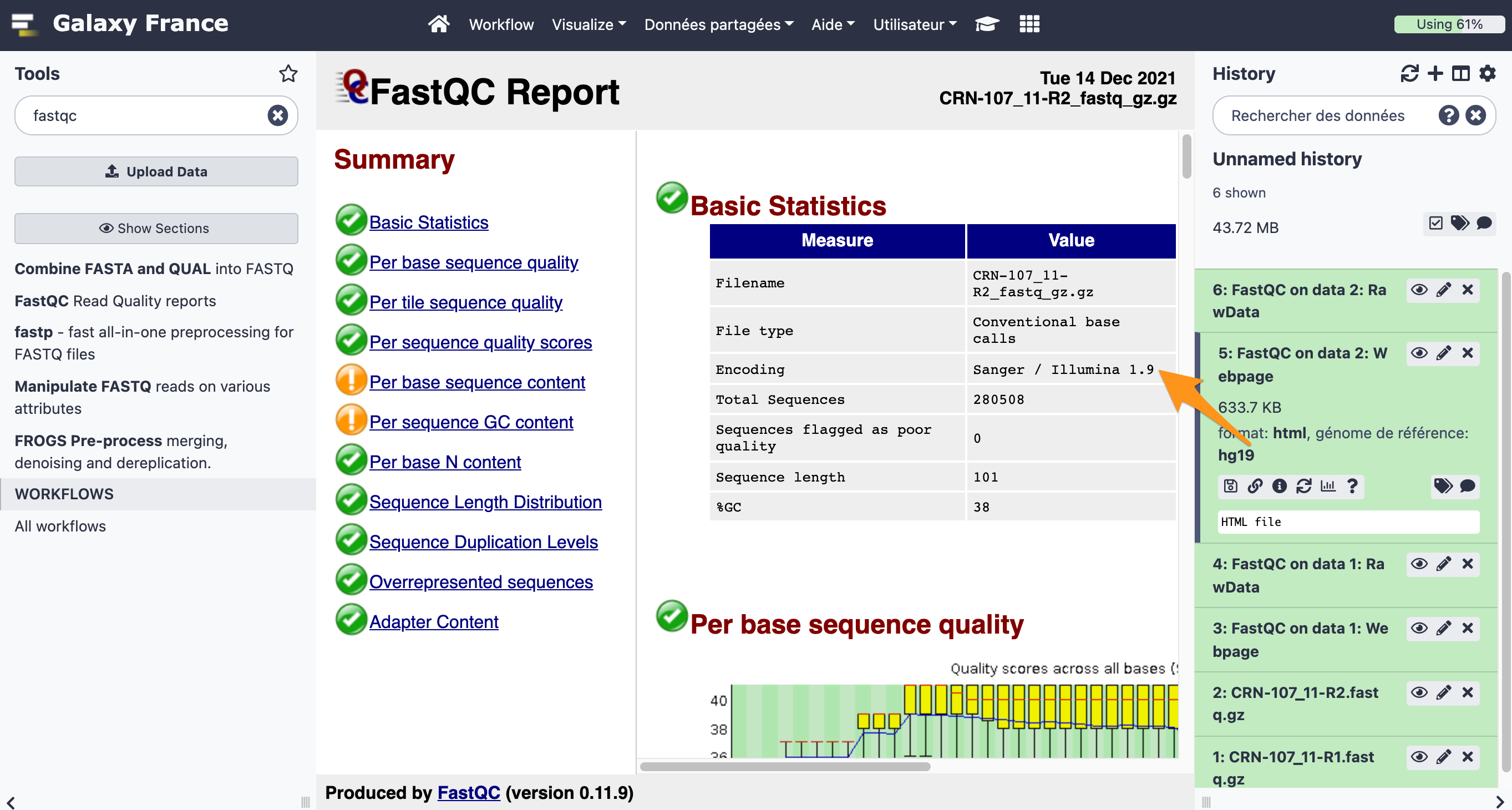
What is the quality encoding of the two fastq files?
15 Running tools without a workflow
Analyze CRN-107 data from reads to variant annotation.
Run the following tools:
- BWA mem to align reads to the reference genome
- Picard markduplicates to identify duplicated reads
- Freebayes to detect variants
- snpEff to annotate variants
To run the tools you will need the following files:
- CRN-107_11-R1.fastq
- CRN-107_11-R2.fastq
- CaptureDesign_chr4.bed (download it from here)
Import missing files from the data library “DNA-seq test datasets”
Here are the parameters to use for each of the tools. All parameters not mentioned are to be used with default values.
- Map with BWA-MEM - map medium and long reads (> 100 bp) against reference genome
- Using reference genome: hg19
- Single or Paired-end reads: Paired
- Select first set of reads: CRN-107_11-R1.fastq
- Select second set of reads: CRN-107_11-R2.fastq.
- Set read groups information? Set read groups (Picard style)
- Read group identifier (ID): Auto-assign Yes
- Read group sample name (SM): Auto-assign Yes
- Library name (LB): Auto-assign Yes
- Platform/technology used to produce the reads (PL): ILLUMINA
- Platform unit (PU): HS026.2
- Sequencing center that produced the read (CN): Genomeast
- Description (DS): CRN-107
- Predicted median insert size (PI): 250
- Date that run was produced (DT): 2017-12-13
- MarkDuplicates examine aligned records in BAM datasets to locate duplicate molecules.
- Select SAM/BAM dataset or dataset collection: output of BWA mem
- Select validation stringency: Silent
- FreeBayes bayesian genetic variant detector
- BAM or CRAM dataset: output (bam) of markduplicates
- Using reference genome: hg19
- Limit analysis to regions in this BED dataset: CaptureDesign_chr4.bed
- SnpEff Variant effect and annotation
- Sequence changes (SNPs, MNPs, InDels): output of GATK Haplotype Caller (VCF)
- Input format: VCF
- Output format: VCF (only if input is VCF)
- Genome source: Downloaded on demand
- Snpff Genome Version Name (e.g. GRCh38.86): hg19
- VCFtoTab-delimited: Convert VCF data into TAB-delimited format
- Select VCF dataset to convert: output of SnpEff
- How many variants are called?
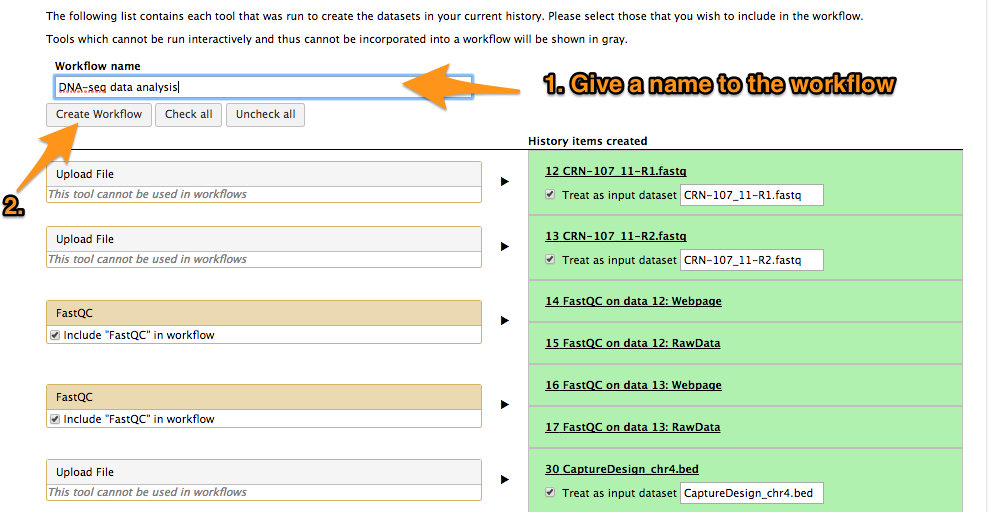
16 Create a workflow out of an existing history
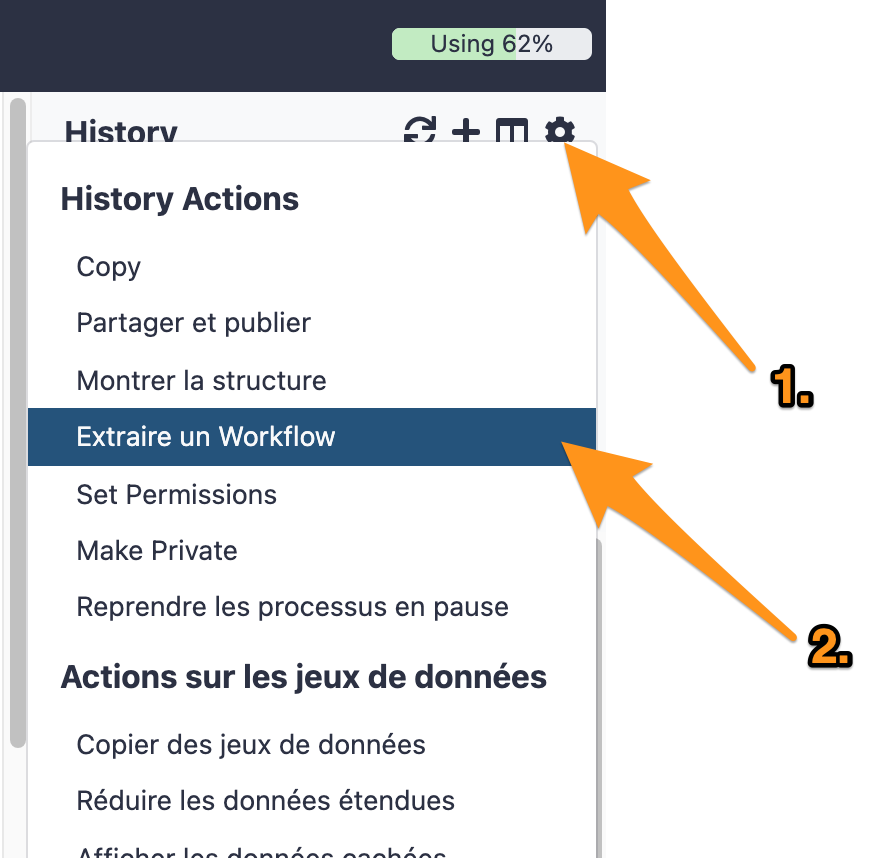
One can create a workflow from an existing history going to the history button and selecting “Extract Workflow”.
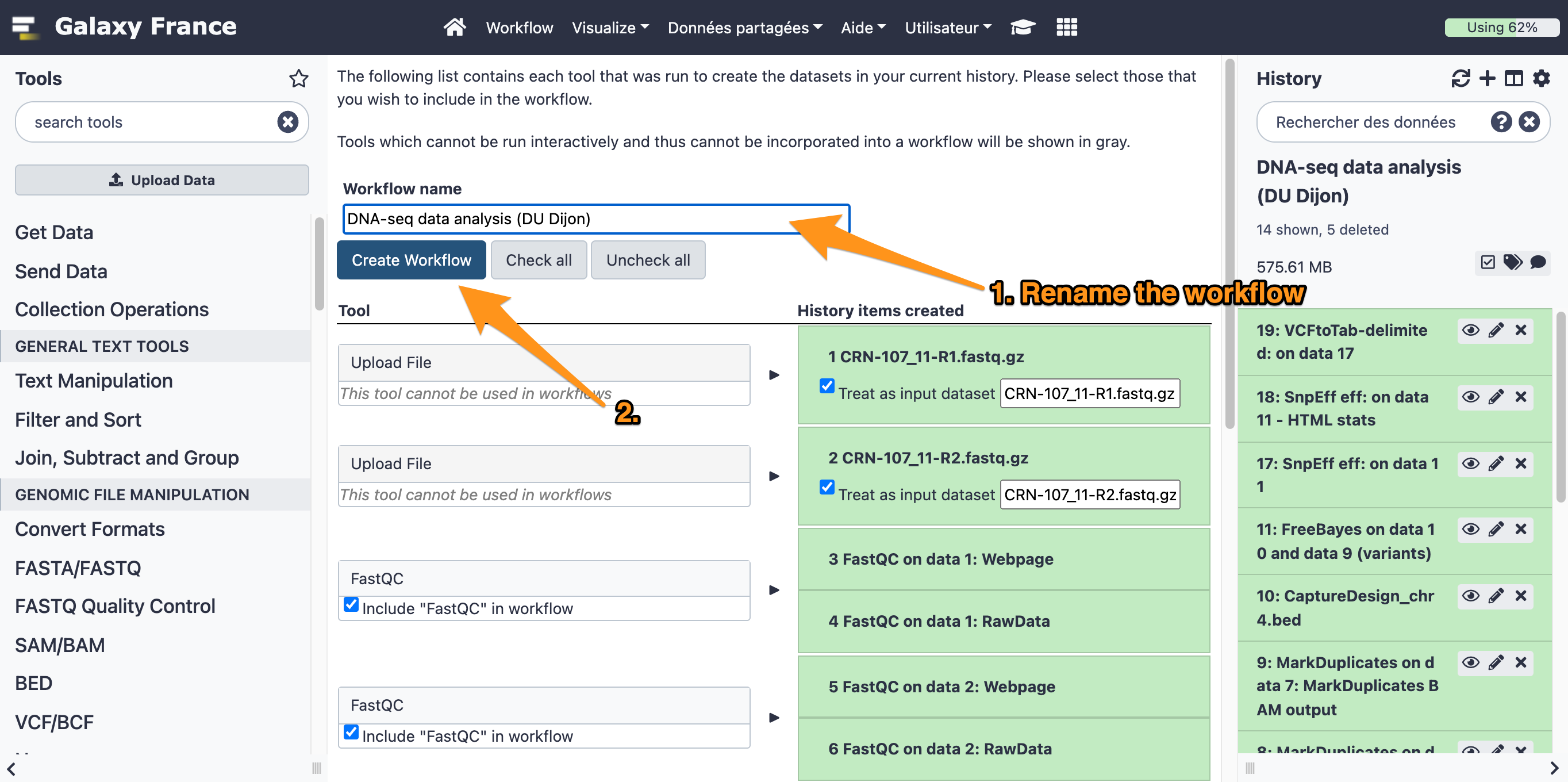
16.1 Extract a workflow out of the history called "DNA-seq data analysis"
16.2 Rename the workflow "DNA-seq data analysis"
17 Edit a workflow with the workflow editor
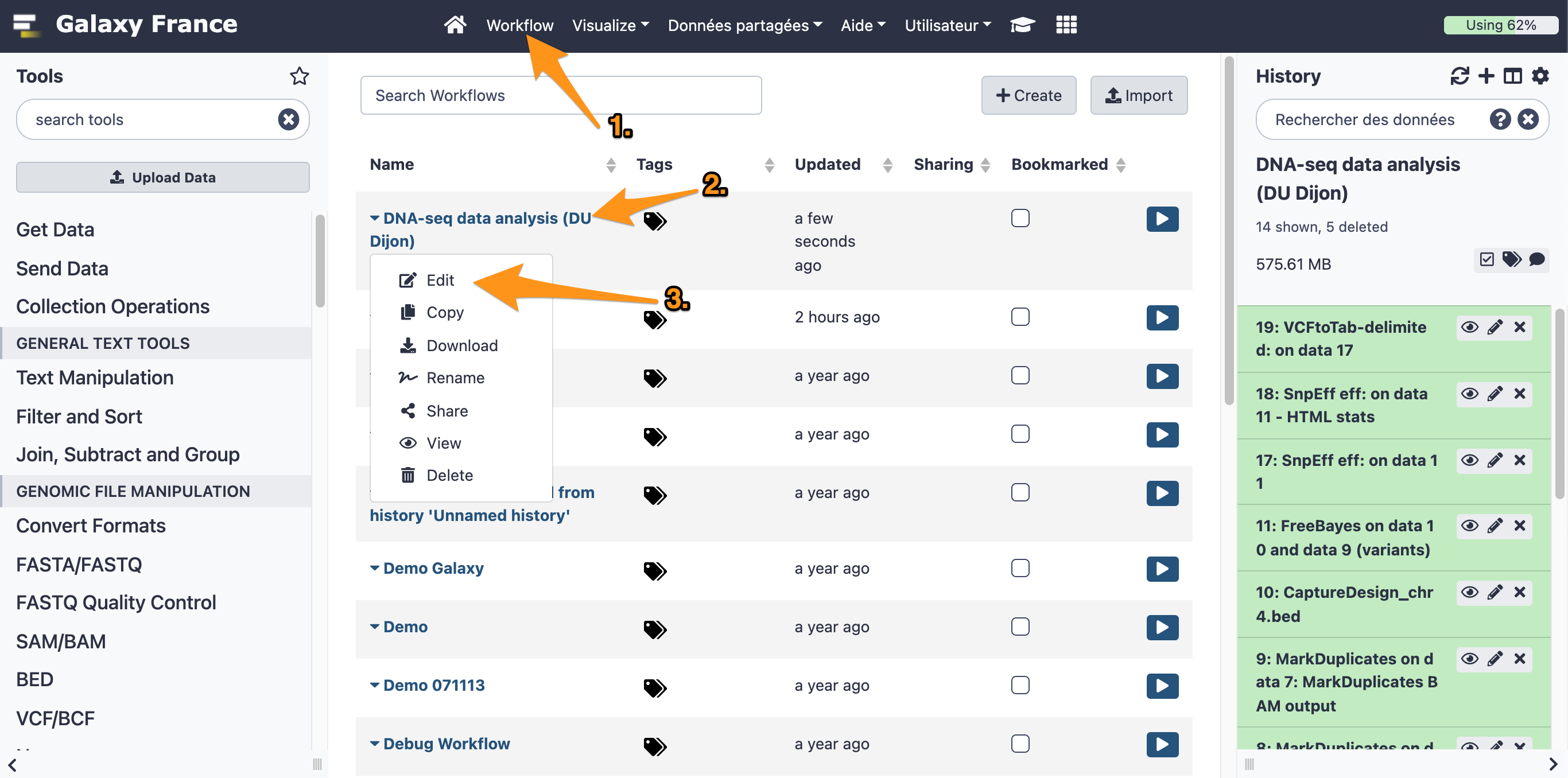
17.1 Open the workflow editor with the workflow "DNA-seq data analysis"
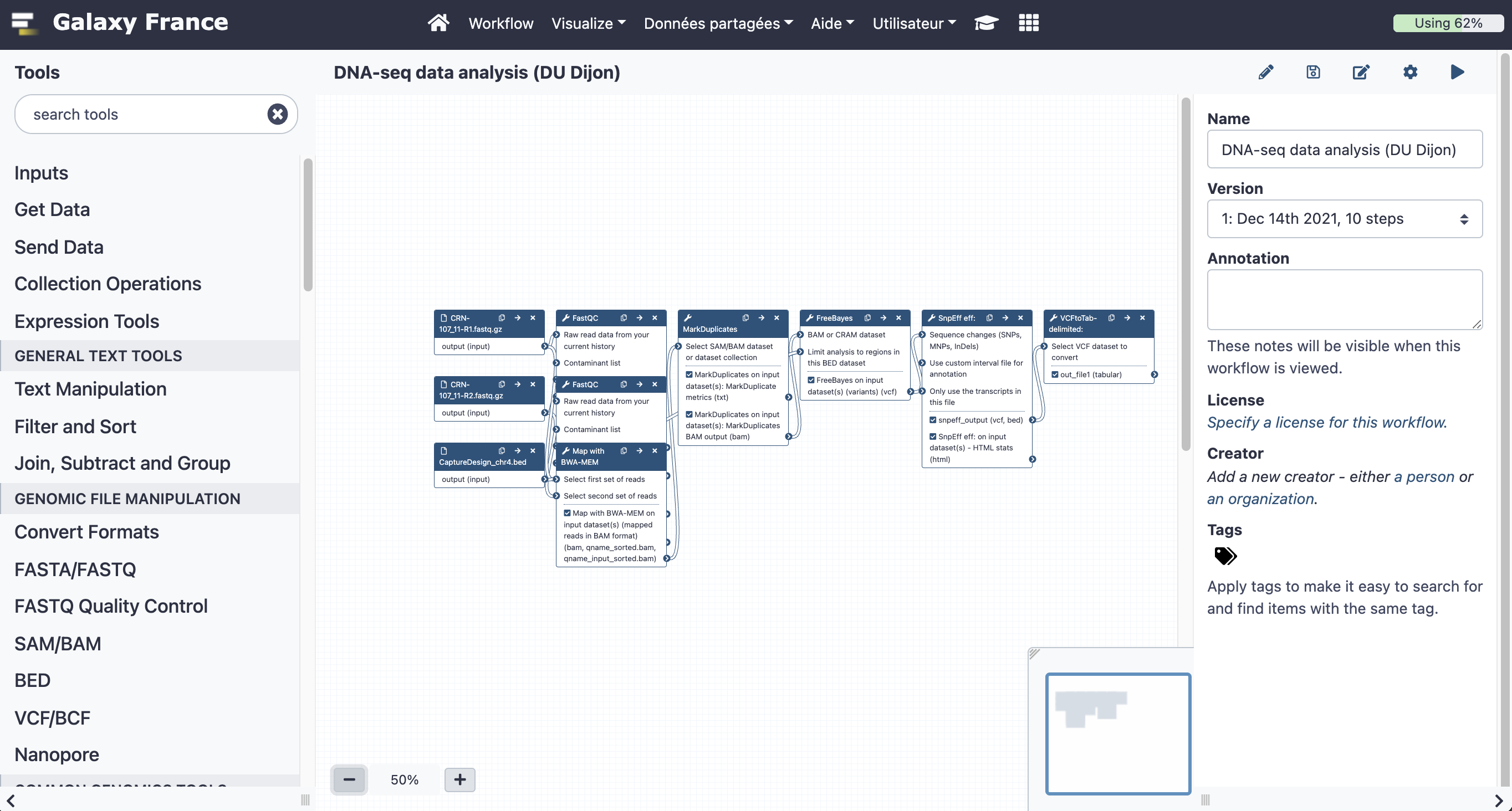
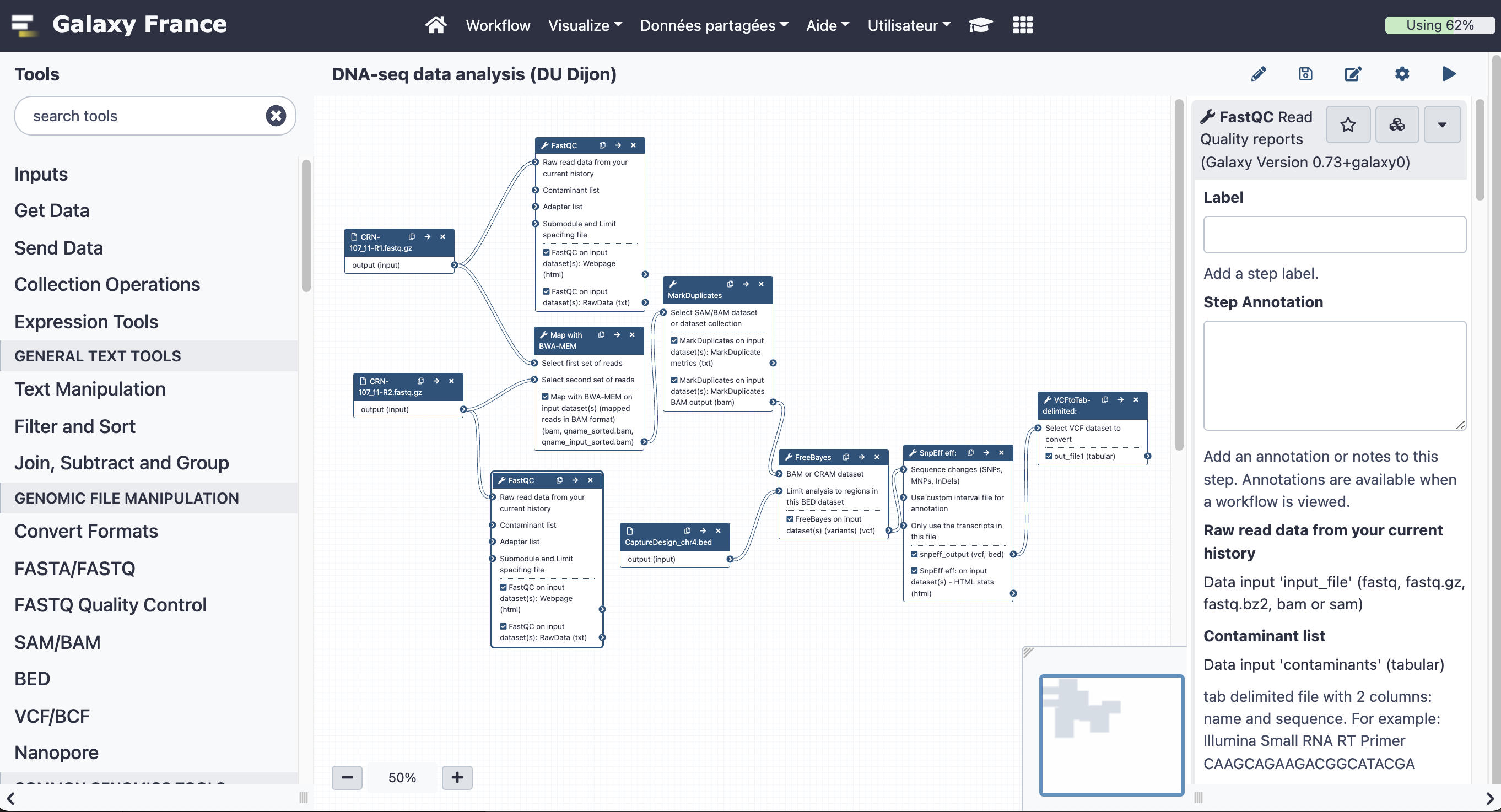
17.2 Add steps to the workflow
Your workflow should look like this before editing:
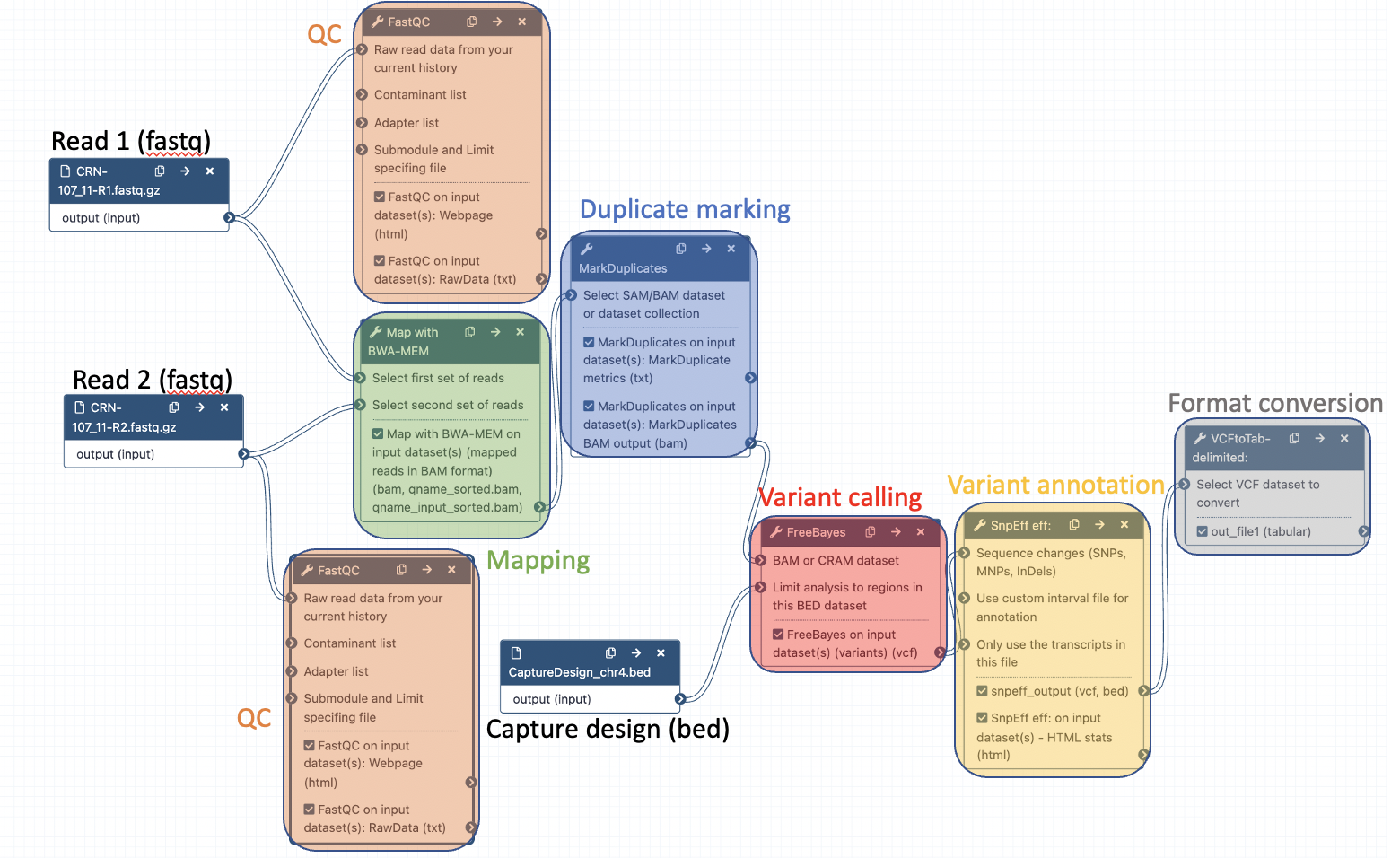
Add the following tools:
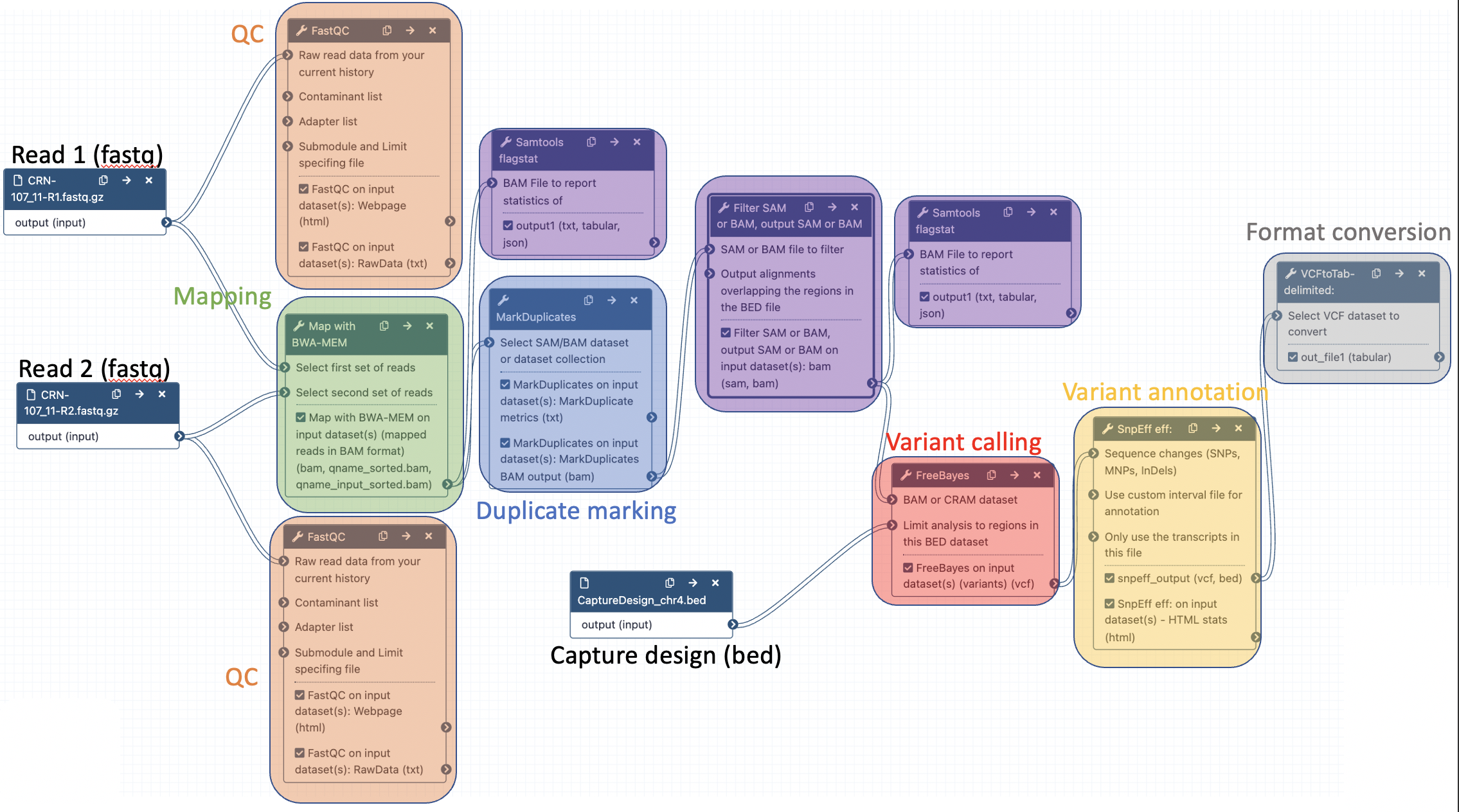
- Samtools flagstat to compute mapping statistics (after BWA mem)
- Filter to select aligned reads with a mapping quality >= 20 (after MarkDuplicates)
- Samtools flagstat to compute mapping statistics after removing reads with low mapping qualities (after Filter)
Here are the parameters to use for each of the tools:
- Flagstat tabulate descriptive stats for BAM dataset
- BAM File to Convert: output of BWA mem
- Filter BAM datasets on a variety of attributes
- BAM dataset(s) to filter: output of Picard MarkDuplicates
- Select BAM property to filter on: mapQuality
- Filter on read mapping quality (phred scale): >=20 (this exact expression, including ”>=”!)
- Flagstat tabulate descriptive stats for BAM dataset
- BAM File to Convert: output of Filter
The final workflow should look like this (new tools are in black boxes):
Save the workflow once you are done editing it:
18 Run a workflow
18.1 Import files
Import the following files from the data library “DNA-seq test datasets” to a new history:
- CRN-107_11-R1.fastq
- CRN-107_11-R2.fastq
- CaptureDesign_chr4.bed
18.2 Run the workflow DNA-seq data analysis
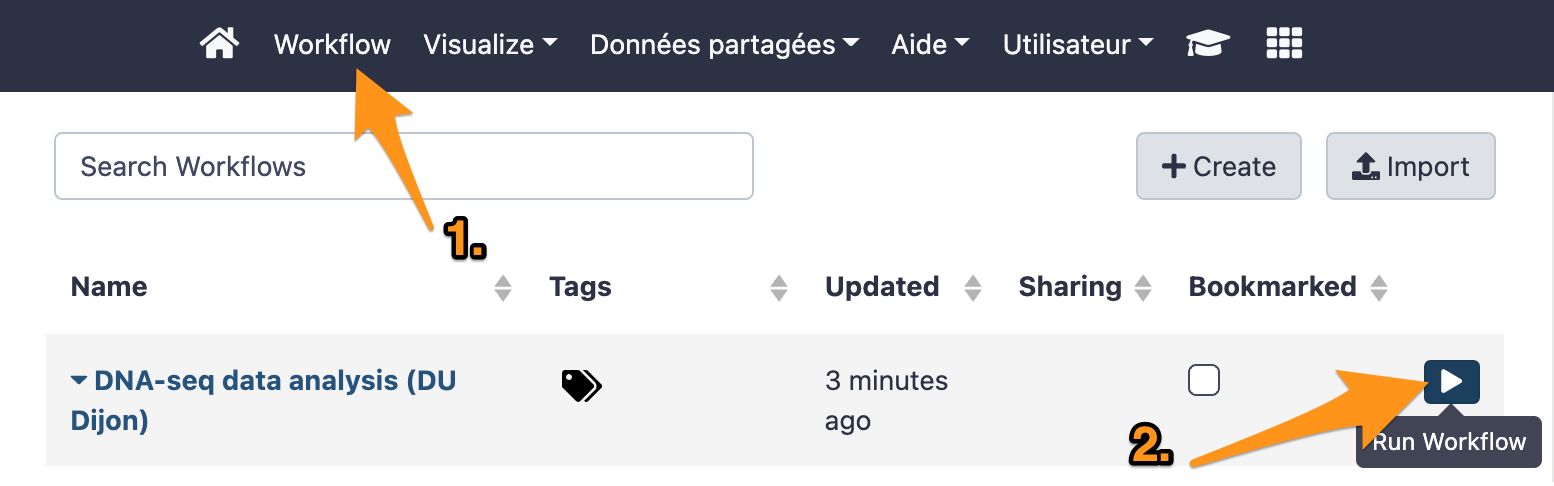
- Choose the right files.
- Check the parameters.
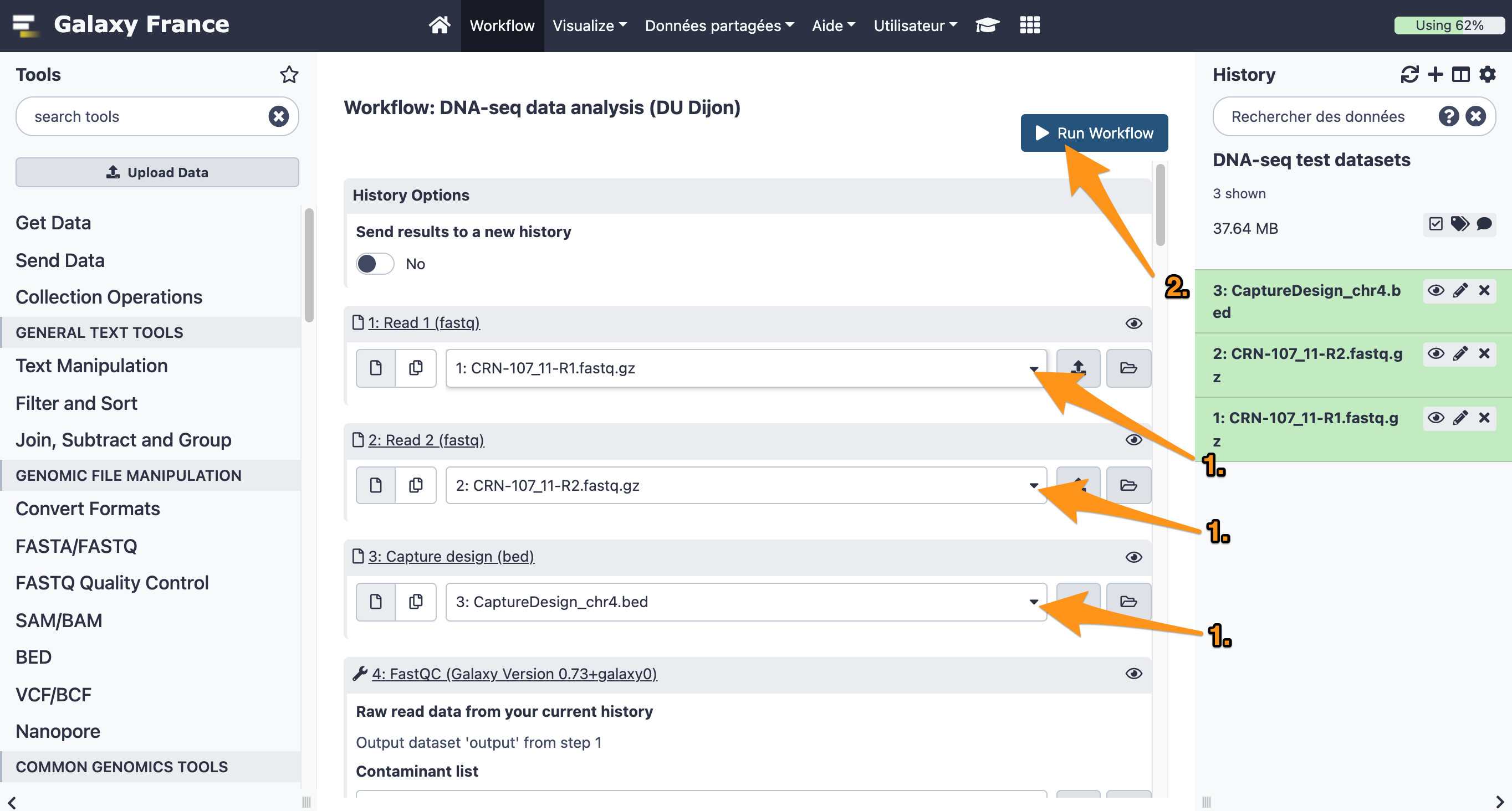
- How many reads are discarded due to the low mapping quality?